Search Results
| نشرة الممارس الصحي | نشرة معلومات المريض بالعربية | نشرة معلومات المريض بالانجليزية | صور الدواء | بيانات الدواء |
|---|
Urago™ tablets contain the active substance febuxostat and are used to
treat gout, which is associated with an excess of a chemical called uric
acid (urate) in the body. In some people, the amount of uric acid builds up
in the blood and may become too high to remain soluble. When this
happens, urate crystals may form in and around the joints and kidneys.
These crystals can cause sudden, severe pain, redness, warmth and
swelling in a joint (known as a gout attack). Left untreated, larger
deposits called tophi may form in and around joints. These tophi may
cause joint and bone damage.
Urago™ works by reducing uric acid levels. Keeping uric acid levels low
by taking Urago™ once every day stops crystals building up, and over
time it reduces symptoms. Keeping uric acid levels sufficiently low for a
long enough period can also shrink tophi.
Urago™ 120mg tablets is also used to treat and prevent high blood levels
of uric acid that may occur when you start to receive chemotherapy for
blood cancers.
When chemotherapy is given, cancer cells are destroyed, and uric acid
levels increase in the blood accordingly, unless the formation of uric acid
is prevented.
Urago™ is for adults.
Do not take Urago™:
• If you are allergic to febuxostat or any of the other ingredients of this
medicine (listed in section 6).
Warnings and precautions
Talk to your doctor before taking Urago™:
• If you have or have had heart failure or heart problems
• If you have or have had renal disease and/or serious allergic reaction to
Allopurinol (amedication used for the treatment of Gout)
• If you have or have had liver disease or liver function test abnormalities
• If you are being treated for high uric acid levels as a result of
Lesch-Nyhan syndrome (a rare inherited condition in which there is too
much uric acid in the blood)
• If you have thyroid problems.
Should you experience allergic reactions to Urago™, stop taking this
medicine (see also section 4). Possible symptoms of allergic reactions
might be:
- rash including severe forms (e.g. blisters, nodules, itchy, exfoliative
rash), itchiness
- swelling of limbs or face
- difficulties in breathing
- fever with enlarged lymph nodes
- but also serious life threatening allergic conditions with cardiac and
circulatory arrest. Your doctor might decide to permanently stop
treatment with Urago™.
There have been rare reports of potentially life-threatening skin rashes
(Stevens-Johnson Syndrome) with the use of Urago™, appearing initially
as reddish target-like spots or circular patches often with central blister on
the trunk. It may also include ulcers in the mouth, throat, nose, genitals
and conjunctivitis (red and swollen eyes). The rash may progress to
widespread blistering or peeling of the skin.
If you have developed Stevens-Johnson Syndrome with the use of
febuxostat, you must not be re- started on Urago™ at any time. If you
develop a rash or these skin symptoms, seek immediate advice from a
doctor and tell that you are taking this medicine.
If you are having a gout attack at the moment (a sudden onset of severe
pain, tenderness, redness, warmth and swelling in a joint), wait for the
gout attack to subside before first starting treatment with Urago™.
For some people, gout attacks may flare up when starting certain
medicines that control uric acid levels. Not everyone gets flares, but you
could get a flare-up even if you are taking Urago™, and especially during
the first weeks or months of treatment. It is important to keep taking
Urago™ even if you have a flare, as Urago™ is still working to lower
uric acid. Over time, gout flares will occur less often and be less painful if
you keep taking Urago™ every day.
Your doctor will often prescribe other medicines, if they are needed, to
help prevent or treat the symptoms of flares (such as pain and swelling in
a joint).
In patients with very high urate levels (e.g. those undergoing cancer
chemotherapy), treatment with uric acid-lowering medicines could lead to
the build-up of xanthine in the urinary tract, with possible stones, even
though this has not been observed in patients being treated with
Urago™ for Tumor Lysis Syndrome.
Your doctor may ask you to have blood tests to check that your liver is
working normally.
Children and adolescents
Do not give this medicine to children under the age of 18 because the
safety and efficacy have not been established.
Other medicines and Urago™
Tell your doctor or pharmacist if you are taking, have recently taken or
might take any other medicines, including medicines obtained without a
prescription.
It is especially important to tell your doctor or pharmacist if you are
taking medicines containing any of the following substances as they may
interact with Urago™ and your doctor may wish to consider necessary
measures:
• Mercaptopurine (used to treat cancer)
• Azathioprine (used to reduce immune response)
• Theophylline (used to treat asthma)
Pregnancy and breast-feeding
It is not known if Urago™ may harm your unborn child.
Urago™ should not be used during pregnancy. It is not known if Urago™
may pass into human breast milk. You should not use Urago™ if you are
breast feeding, or if you are planning to breastfeed.
If you are pregnant or breast-feeding, think you may be pregnant or are
planning to have a baby, ask your doctor or pharmacist for advice before
taking this medicine.
Driving and using machines
Be aware that you may experience dizziness, sleepiness, blurred vision
and numbness or tingling sensation during treatment and should not drive
or operate machines if affected.
Urago™ contains lactose
Urago™ tablets contain lactose (a type of sugar). If you have been told
that you have an intolerance to some sugars contact your doctor before
taking this medicine.
Always take this medicine exactly as your doctor has told you. Check
with your doctor or pharmacist if you are not sure.
• The usual dose is one tablet daily. The back of the blister pack is marked
with the days of the week to help you check that you have taken a dose
each day.
• The tablets should be taken by mouth and can be taken with or without
food.
Gout
Urago™ is available as either an 40mg tablet or a 80mg tablet. Your
doctor will have prescribed the strength most suitable for you.
Continue to take Urago™ every day even when you are not experiencing
gout flare or attack.
Prevention and treatment of high uric acid levels in patients
undergoing cancer chemotherapy
Urago™ is available as a 40mg and 80mg tablet.
Start taking Urago™ two days before chemotherapy and continue its use
according to your doctor‟s advice. Usually treatment is short-term.
If you take more Urago™ than you should
In the event of an accidental overdose ask your doctor what to do, or
contact your nearest accident and emergency department.
If you forget to take Urago™
If you miss a dose of Urago™ take it as soon as you remember unless it is
almost time for your next dose, in which case miss out the forgotten dose
and take your next dose at the normal time. Do not take a double dose to
make up for a forgotten dose.
If you stop taking Urago™
Do not stop taking Urago™ without the advice of your doctor even if you
feel better. If you stop taking Urago™ your uric acid levels may begin to
rise and your symptoms may worsen due to the formation of new crystals
of urate in and around your joints and kidneys.
If you have any further questions on the use of this medicine, ask your
doctor or pharmacist.
Like all medicines, this medicine can cause side effects, although not
everybody gets them.
Stop taking this medicine and contact your doctor immediately or go to an
emergency department nearby if the following rare (may affect up to 1 in
1,000 people) side effects occur, because a serious allergic reaction might
follow:
• anaphylactic reactions, drug hypersensitivity (see also section 2
“Warnings and precautions”)
• potentially life-threatening skin rashes characterised by formation of
blisters and shedding of the skin and inner surfaces of body cavities, e.g.
mouth and genitals, painful ulcers in the mouth and/or genital areas,
accompanied by fever, sore throat and fatigue (Stevens- Johnson
Syndrome/ Toxic Epidermal Necrolysis), or by enlarged lymph nodes,
liver enlargement, hepatitis (up to liver failure), raising of the white-cells
count in the blood (drug reaction with eosinophilia and systemic
symptoms-DRESS) (see section 2)
• generalised skin rashes
The common side effects (may affect up to 1 in 10 people) are:
• abnormal liver test results
• diarrhoea
• headache
• rash (including various types of rash, please see below under
“uncommon” and “rare” sections)
• nausea
• increase in gout symptoms
• localised swelling due to retention of fluids in tissues (oedema)
Other side effects which are not mentioned above are listed below.
Uncommon side effects (may affect up to 1 in 100 people) are:
• decreased appetite, change in blood sugar levels (diabetes) of which a
symptom may be excessive thirst, increased blood fat levels, weight
increase
• loss of sex drive
• difficulty in sleeping, sleepiness
• dizziness, numbness, tingling, reduced or altered sensation
(hypoaesthesia, hemiparesis or paraesthesia), altered sense of taste,
diminished sense of smell (hyposmia)
• abnormal ECG heart tracing, irregular or rapid heartbeats, feeling your
heart beat (palpitation)
• hot flushes or flushing (e.g. redness of the face or neck), increased
blood pressure, bleeding (hemorrhage, seen only in patients taking
chemotherapy for blood disorders)
• cough, shortness of breath, chest discomfort or pain, inflammation of
nasal passage and/or throat (upper respiratory tract infection), bronchitis
• dry mouth, abdominal pain/discomfort or wind, heartburn/indigestion,
constipation, more frequent passing of stools, vomiting, stomach
discomfort
• itching, hives, skin inflammation, skin discoloration, small red or purple
spots on the skin, small, flat red spots on the skin, flat, red area on the
skin that is covered with small confluent bumps, rash, areas of redness
and spots on the skin, other type of skin conditions
• muscle cramp, muscle weakness, pain/ache in muscles/joints, bursitis or
arthritis (inflammation of joints usually accompanied by pain, swelling
and/or stiffness), pain in extremity, back pain, muscle spasm
• blood in the urine, abnormal frequent urination, abnormal urine tests
(increased level of proteins in the urine), a reduction in the ability of the
kidneys to function properly
• fatigue, chest pain, chest discomfort
• stones in the gallbladder or in bile ducts (cholelithiasis)
• increase in blood thyroid stimulating hormone (TSH) level
• changes in blood chemistry or amount of blood cells or platelets
(abnormal blood test results)
• kidney stones
• erectile difficulties
Rare side effects (may affect up to 1 in 1,000 people) are:
• muscle damage, a condition which on rare occasions can be serious. It
may cause muscle problems and particularly, if at the same time, you feel
unwell or have a high temperature it may be caused by an abnormal
muscle breakdown. Contact your doctor immediately if you experience
muscle pain, tenderness or weakness
• severe swelling of the deeper layers of the skin, especially around the
lips, eyes, genitals, hands, feet or tongue, with possible sudden difficult
breathing
• high fever in combination with measles-like skin rash, enlarged lymph
nodes, liver enlargement, hepatitis (up to liver failure), raising of the
white-cells count in the blood (leukocytosis, with or without eosinophilia)
• reddening of the skin (erythema), rash in various types (e.g. itchy, with
white spots, with blisters, with blisters containing pus, with shedding of
the skin, measles-like rash), widespread erythema, necrosis, and bullous
detachment of the epidermis and mucous membranes, resulting in
exfoliation and possible sepsis (Stevens-Johnson Syndrome/Toxic
epidermal necrolysis)
• nervousness
• feeling thirsty
• vringing in the ears
• blurred vision, change in vision
• hair loss
• mouth ulceration
• inflammation of the pancreas: common symptoms are abdominal pain,
nausea and vomiting
• increased sweating
• weight decrease, increased appetite, uncontrolled loss of appetite
(anorexia)
• muscle and/or joint stiffness
• abnormally low blood cell counts (white or red blood cells or platelets)
• urgent need to urinate
• changes or decrease in urine amount due to inflammation in the kidneys
(tubulointerstitial nephritis)
• inflammation of the liver (hepatitis)
• yellowing of the skin (jaundice)
• liver damage
• increased level of creatine phosphokinase in blood (an indicator of
muscle damage)
Reporting of side effects
If you get any side effects, talk to your doctor or pharmacist. This
includes any possible side effects not listed in this leaflet. By reporting
side effects you can help provide more information on the safety of this
medicine.
Keep out of the sight and reach of children.
Do not use this medicine after the expiry date, which is stated on the
packaging.
Do not store above 30 °C.
Do not throw away any medicines via wastewater or household waste.
Ask your pharmacist how to throw away medicines you no longer use.
These measures will help protect the environment.
What Urago™ contains
The active substance is Febuxostat.
Each Film Coated Tablet contains 40mg or 80mg of Febuxostat.
The other ingredients are:
Core: Lactose Monohydrate, Microcrystalline Cellulose, Hydroxy propyl
Cellulose, Colloidal Silicon Dioxide, Croscarmellose Sodium and
Magnesium stearate.
Coating: Polyvinyl alcohol, Macrogol/PEG, Talc, Quinoline yellow,
Brilliant blue, Sunset yellow and Titanium dioxide
Jamjoom Pharmaceuticals Co., Makkah Region, Jeddah, Saudi Arabia.
Tel: +966-12-6081111, Fax: +966-12-6081222
Website: www.jamjoompharma.com
To report any side effect(s):
• Saudi Arabia:
The National Pharmacovigilance and Drug Safety Centre (NPC)
o Fax: +966-11-205-7662
o Call NPC at +966-11-2038222, Ext: 2317-2356-2340.
o Reporting hotline: 19999
o E-mail: npc.drug@sfda.gov.sa
o Website: www.sfda.gov.sa/npc
• Other GCC States:
− Please contact the relevant competent authority.
راص على المادة الفعّالة فیبوكسوستات ویستخدم لعلاج مرض النقرس ™ یحتوي عقار یوراجو
المصاحب لزیادة مادة كیمیائیة تُسمى حمض الیوریك (الیورات) في الجسم. حیث یتراكم حمض
الیوریك في الدم لدى بعض المرضى وقد تصبح نسبتھ مرتفعة جدًا مما یجعلھ غیر قابلاً للذوبان.
عند حدوث ذلك، قد تتكون بلورات الیورات داخل وحول المفاصل والكلى. حیث قد تتسبب ھذه
البلورات في حدوث ألم حاد ومفاجئ واحمرار وحرارة وتورم المفاصل (ویُسمى ذلك بنوبة
النقرس). إذا لم یتم معاجلة ھذه الحالة، فقد تتكون رواسب رملیة كبیرة تُسمى (تُوَف) داخل
وحول المفاصل. وقد تُسبب ھذه الرواسب الرملیة (التُوَف) حدوث تلف في المفاصل والعظام.
على خفض مستویات حمض الیوریك. حیث یؤدي بقاء مستویات حمض ™ یعمل عقار یوراجو
مرة یومیًا إلى وقف تراكم البلورات وتخفیف ™ الیوریك منخفضًا بفضل تناول عقار یوراجو
أعراضھ بمرور الوقت. كما یساعد بقاء نسبة حمض الیوریك منخفضة بشكل كافي لفترة طویلة
وكافیة على قلة الرواسب الرملیة"التوف".
۱۲۰ ملجم أقراص أیضًا لعلاج ارتفاع مستویات حمض الیوریك في الدم ™ یُتناول عقار یوراجو
وللوقایة من الإصابة بھ والذي قد یحدث عند البدء في تلقي العلاج الكیمیائي لمعالجة سرطان
الدم.
یتم تدمیر الخلایا السرطانیة عند تلقي العلاج الكیمیائي، وبالتالي، ترتفع مستویات حمض الیوریك
في الدم، ما لم یتم منع تكون حمض الیوریك.
لعلاج المرضى من البالغین. ™ یستخدم عقار یوراجو
۲. ما الذي تحتاج إلى معرفتھ قبل تناوُل عقار یوراجو
في الحالات الآتیة: ™ لا تتناول عقار یوراجو
• إذا كنت تعاني من حساسیة تجاه فیبوكسوستات أو أي مكون من المكونات الأخرى الداخلة في
.( تركیب ھذا الدواء (المُدرجة في القسم رقم ٦
تحذیرات واحتیاطات
في الحالات الآتیة: ™ تحدّث إلى الطبیب المعالج لك قبل تناول عقار یوراجو
• إذا كنت تُعاني أو قد عانیت من فشل القلب أو مشاكل بالقلب.
• إذا كنت تعاني أو قد عانیت من مرض كلوي و/أو تفاعلات حساسیة خطیرة تجاه عقار
ألوبیورینول (دواء یستخدم لعلاج النقرس)
• إذا كنت تُعاني أو قد عانیت مسبقًا من مرض بالكبد أو ظھور نتائج غیر طبیعیة في فحص
وظائف الكبد.
• إذا كنت تتلقي دواءً لعلاج ارتفاع مستویات حمض الیوریك في الدم نتیجة لمتلازمة لیش-نیھان
(حالة وراثیة نادرة یرتفع فیھا مستوى حمض الیوریا في الدم)
• إذا كنت تعاني من مشاكل في الغدة الدرقیة.
توقف عن تناول ھذا العقار (انظر أیضًا ،™ في حالة حدوث تفاعلات حساسیة تجاه عقار یوراجو
القسم رقم ٦). وقد تتمثل أعراض تفاعلات الحساسیة المحتملة فیما یلي:
- طفح جلدي بما في ذلك بعض أشكال الطفح الجلدي الحاد (على سبیل المثال ظھور بثور،
تكتلات، حكة، طفح جلدي تقشري)، حكة
- تورم بالأطراف أو الوجھ.
- صعوبات في التنفس.
- حمى مصاحبة لتضخم العقد اللیمفاویة.
- أیضًا حالات حساسیة تھدد الحیاة بالخطر مصحوبة بحالة توقف القلب وإضراب الدورة
.™ الدمویة. قد یقرر الطبیب المعالج لك التوقف نھائیًا عن تناول عقار یوراجو
تم الإبلاغ عن احتمال حدوث حالات طفح جلدي تھدد الحیاة بالخطر (متلازمة ستیفنز جونسون)
التي تظھر في بدایة الأمر كبقع دائریة حمراء أو بقع دائریة ™ بسبب تناول عقار یوراجو
مصحوبة غالبًا ببثور متمركزة على الجسم. وقد تتمثل أیضًا في وجود تقرحات بالفم والحلق
والأنف والأعضاء التناسلیة والتھاب الملتحمة (احمرار وتورّم العین). كما قد یتفاقم الطفح الجلدي
ویتحول إلى بثور منتشرة أو تقشر الجلد.
مرة أخرى إذا أُصبت بمتلازمة ستیفنز جونسون ™ یُحظر علیك إعادة استخدام عقار یوراجو
أثناء استخدام فیبوكسوستات. وفي حالة تفاقم حالة الطفح الجلدي أو ھذه الأعراض الجلدیة،
یُرجى استشارة الطبیب المعالج لك فورًا وإخباره بأنك تتناول ھذا الدواء.
إذا كنت تعاني من نوبة النقرس في الوقت الحالي (بدایة حدوث ألم حادّ ومفاجئ، ضیق وألم،
احمرار، حرارة وتورم في المفاصل)، فانتظر زوال نوبة النقرس قبل البدء في تناول عقار
.™ یوراجو
قد تتفاقم حدة نوبات النقرس في بعض المرضى عند البدء في تناول أدویة معینة للتحكم في
مستویات حمض الیوریك. ولكن لا یحدث ذلك لدى جمیع المرضى، ولكنك قد تعاني من تفاقم حدة
وخصوصًا أثناء الأسابیع أو الأشھر الأولى ،™ نوبة النقرس حتى إذا كنت تتناول عقار یوراجو
حتى إذا كنت ™ لتناول العلاج بھذا الدواء. من المھم جدًا الاستمرار في تناول عقار یوراجو
في خفض مستوى حمض ™ تعاني من تفاقم حدة نوبات النقرس، حیث یستمر عقار یوراجو
الیوریك. وبمرور الوقت، سیقل تعرضك لحدوث نوبات النقرس كما ستكون النوبات أقل ألمًا إذا
یومیًا. ™ ما استمریت في تناول عقار یوراجو
غالبًا ما سیقوم الطبیب المعالج لك بوصف أدویة أخرى، إذا لزم الأمر، للوقایة من تفاقم نوبات
النقرس وعلاج أعراضھا (مثل الشعور بألم وورم في المفاصل).
بالنسبة للمرضى الذین یعانون من ارتفاع شدید في مستویات الیورات (مثل المرضى الذین
یخضعون لعلاج السرطان الكیمیائي)، فقد یؤدي العلاج بالأدویة المخفضة لحمض الیوریك في
الدم إلى تراكم الزانثین في الجھاز البولي، مع احتمال تكون حصوات، على الرغم من عدم
لعلاج متلازمة انحلال الورم. ™ ملاحظة ذلك في المرضى الذین یتناولون عقار یوراجو
قد یُقرر الطبیب المعالج لك أیضًا إجراء فحوصات للدم للتحقق من أن الكبد لدیك یعمل بشكل
طبیعي.
الأطفال والمراھقین
لا یستخدم ھذا الدواء في المرضى من الأطفال والمراھقین الذین تقل أعمارھم عن ۱۸ عامًا؛ إذ
لم یثبت أمان وفعالیة تناول ھذا الدواء في ھذه الفئة العمریة.
مع أدویة أخرى ™ تناول عقار یوراجو
أخبر الطبیب المعالج لك أو الصیدلي الخاص بك إذا كنت تتناول أو تناولت مؤخرًا أو قد تتناول
أیَّة أدویة أخرى بما في ذلك الأدویة التي حصلت علیھا بدون وصفة طبیة.
من المھم جدَا إخبار الطبیب المعالج لك أو الصیدلي الخاص بك إذا كنت تتناول أدویة تحتوي
وقد یرغب الطبیب المعالج لك ™ على المواد التالیة حیث قد تتفاعل ھذه المواد مع عقار یوراجو
في اتخاذ التدابیر اللازمة:
• مركابتوبورین (یُستَخدَم لعلاج مرض السرطان).
• آزاثیوبرین (یُستَخدَم لخفض الاستجابة المناعیة).
• تیوفیلین (یُستَخدَم لعلاج الربو).
الحمل والرضاعة الطبیعیة
یضر بالجنین أم لا. ولكن یُحظر تناول عقار ™ من غیر المعروف ما إذا كان عقار یوراجو
یُفرز في لبن الأم أم ™ أثناء فترة الحمل. من غیر المعروف ما إذا كان عقار یوراجو ™ یوراجو
إذا كنتِ تمارسین الرضاعة الطبیعیة، أو إذا كنتِ تنوین ™ لا. لذلك یُحظر تناول عقار یوراجو
إرضاع طفلك طبیعیًا.
إذا كُنتِ حاملًا أو تمارسین الرضاعة الطبیعیة أو تعتقدین أنكِ قد تكونین حاملًا أو تخططین
لإنجاب طفل، فاستشیري الطبیب المعالج لكِ أو الصیدلي الخاص بكِ قبل تناوُل ھذا الدَّواء.
القیادة واستخدام الآلات
یُرجى العلم باحتمال الإصابة بدوخة ونعاس وعدم وضوح الرؤیة وشعور بتنمیل أو وخز أثناء
فترة العلاج، وینبغي عدم القیادة أو استخدام الآلات إذا عانیت من أيّ من الأعراض السابقة.
على اللاكتوز ™ یحتوي عقار یوراجو
أقراص على اللاكتوز (نوع من السكر). لذلك قمْ بالاتصال بالطبیب ™ یحتوي عقار یوراجو
المعالج لك قبل تناول ھذا الدَّواء إذا قد تم إخبارك بأنك لا تتحمل بعض أنواع السكریات.
تناول دائمًا ھذا الدَّواء تمامًا كما أخبرك الطبیب المعالج لك. استشر الطبیب المعالج لك أو
الصیدلي الخاص بك إذا لم تكن متأكدًا من كیفیة الاستخدام.
• الجرعة المعتادة ھي قرص واحد یومیًا. حیث یكون مكتوبًا على ظھر الشریط أیام الأسبوع
للمساعدة في التأكد من تناولك الجرعة كل یوم.
• یجب تناول الأقراص عن طریق الفم ویمكن تناولھا مع الطعام أو بدونھ.
في حالة علاج النّقرس
إمّا في شكل ٤۰ ملجم أقراص أو على شكل قرص ۸۰ ملجم أقراص. ™ یتوفر عقار یوراجو
سیصف الطبیب المعالج لك التركیز الأكثر ملائمةً لحالتك الصحیة.
یومیًا حتى إذا لم تعاني من نوبات النقرس أو تفاقم ™ یجب الاستمرار في تناول عقار یوراجو
تلك النوبات.
في حالة الوقایة ارتفاع مستویات حمض الیوریك وعلاجھ في المرضى الذین یتلقون علاج
السرطان الكیمیائي
في شكل ٤۰ ملجم و ۸۰ ملجم أقراص. ™ یتوفر عقار یوراجو
قبل تلقي العلاج الكیمیائي بیومین واستمر في تناولھ حسب إرشادات ™ ابدأ تناولَ عقارِ یوراجو
الطبیب المعالج لك. عادةً ما یكون العلاج قصیر المدى.
:™ إذا تناولت كمیة أكثر مما یجب من عقار یوراجو
في حالة تناول جرعة زائدة عن طریق الخطأ، استشر الطبیب المعالج عما یجب علیك اتباعھ أو
اتصل بأقرب قسم للحوادث والطوارئ.
:™ إذا أغفلت تناول عقار یوراجو
فتناولھا بمجرد تذكرك لھا ما لم یكن قد حان وقت ،™ إذا أغفلت تناول جرعة من عقار یوراجو
تناول الجرعة التَّالیة. في حالة تجاوز الجرعة المنسیة وحلول موعد الجرعة التالیة، فتناول
الجرعة التالیة في وقتھا المعتاد، ولا تتناول جرعة مضاعفة لتعویض جرعة أغفلتھا.
:™ إذا توقفت عن تناول عقار یوراجو
بدون استشارة الطبیب المعالج لك حتى إذا شعرت بتحسّن. ™ لا تتوقف عن تناوُل عقار یوراجو
فقد تبدأ مستویات حمض الیوریك في الارتفاع وربما ،™ إذا توقفت عن تناول عقار یوراجو
تعاني من تفاقم الأعراض نتیجة لتكون جدید لبلورات الیورات في المفاصل والكلى وحولھا.
استشر الطبیب المعالج لك أو الصیدلي الخاص بك إذا كانت لدیك أیّة أسئلة إضافیة حول تناول
ھذا الدواء.
قد یُسبب ھذا الدواء، مثلھ مثل كافة الأدویة، آثارًا جانبیة، على الرغم من عدم حدوثھا لجمیع
المرضى.
توقف عن تناول ھذا الدواء واتصل بالطبیب المعالج لك فورًا أو توجھ إلى أقرب قسم طوارئ إذا
عانیت من أي من الآثار الجانبیة التالیة التي تحدث في حالات نادرة (قد تؤثر على ما یصل إلى
مریض واحد من بین كل ۱۰۰۰ مریض)، لأنھ قد یعقب ذلك تفاعلات حساسیة شدیدة:
• تفاعلات تأقیة، حساسیة مفرطة تجاه العقار (انظر أیضًا القسم رقم ۲ "تحذیرات واحتیاطات").
• احتمال حدوث حالات طفح جلدي تھدد الحیاة مصحوبة بتكون بثور ونزیف الجلد والأسطح
الداخلیة لتجاویف الجسم مثل الفم والأعضاء التناسلیة، تقرحات مؤلمة في الفم و/أو مناطق
الأعضاء التناسلیة مصحوبة بحُمى والتھاب الحلق وشعور بالتعب والإجھاد (متلازمة ستیفنز
جونسون/ تَقَشُّرُ الأَنْسِجَةِ المُتَمَوِّتَةِ البَشْرَوِیَّةِ التَّسَمُّمِيّ) أو مصحوبة بتضخم العقد اللیمفاویة،
تضخم الكبد، التھاب الكبد (وصولًا إلى فشل الكبد)، ارتفاع عدد خلایا الدم البیضاء بالدّم (تفاعل
("DRESS " الدواء مع فرط الكریات الحمضیة وأعراض الأمراض الجھازیة
( (انظر القسم رقم ۲
• حالات طفح جلدي معمّم (منتشر في جمیع أجزاء الجسم).
آثار جانبیة شائعة (قد تُؤثر على ما یصل إلى مریض واحد من بین كل ۱۰ مرضى) تشمل ما
یلي:
• ظھور نتائج غیر طبیعیة في فحوصات الكبد.
• إسھال.
• صداع.
• طفح جلدي (یتضمن أنواع مختلفة من الطفح الجلدي، یُرجى النظر في قسمي "آثار جانبیة غیر
شائعة"، و"آثار جانبیة نادرة").
• غثیان.
• زیادة أعراض النقرس.
• تورم موضعي نتیجة لاحتباس السوائل في الأنسجة (وذمة).
الآثار الجانبیة غیر المدرجة أعلاه، یتم ذكرھا فیما یلي.
آثار جانبیة غیر شائعة (قد تُؤثر على ما یصل إلى مریض واحد من بین كل ۱۰۰ مریض):
• انخفاض الشھیة أو تغیر مستویات السكر في الدم (مرض السكّري) الذي قد یصحبھ أعراض
مثل شدة العطش، زیادة مستویات الدھون في الدم، زیادة الوزن.
• فقدان الرغبة الجنسیة.
• صعوبة في النوم، نُعاس.
• دوخة أو تنمیل أو وخز أو تغیر أو انخفاض في حاسة اللمس (ضعف الإحساس، شلل نصفي أو
تنمیل)، تغیر في حاسة الذوق، تدھور في حاسة الشم (ضعف حاسة الشم).
• اضطراب في تتبع الإشارات الكھربیة للقلب، ضربات قلب سریعة وغیر منتظمة، شعور بعدم
انتظام ضربات القلب (خفقان القلب)
• ھبّات ساخنة أو احمرار (مثل احمرار الوجھ أو الرقبة)، ارتفاع ضغط الدم، (نزیف، یحدث
فقط بالنسبة للمرضى الذین یتلقون علاجًا كیمیائیًا لعلاج اضطرابات الدم).
• سعال، ضیق التنفس، ضیق أو ألم بالصدر، التھاب الممر الأنفي و/أو الحَلْق (عدوى الجھاز
التنفسي العلوي)، التھاب الشعب الھوائیة.
• جفاف الفَم، ألم بالبطن/ اضطراب أو غازات، حموضة المعدة/عسر الھضم، إمساك، كثرة
التبرز، قيء، شعور بعدم ارتیاح المعدة.
• حكة، شرى، التھاب الجلد، تغیر لون الجلد، بقع حمراء أو أرجوانیة صغیرة على الجلد، بقع
حمراء سطحیة صغیرة على الجلد، مناطق حمراء سطحیة على الجلد مغطاة بحبوب غزیرة
وصغیرة، طفح، مناطق حمراء وبقع على الجلد، وأنواع أخرى من الحالات الجلدیة.
• تقلصات عضلیة، ضعف العضلات، ألم/ وجع في العضلات/ المفاصل، التھاب الجراب "ھي
حالة مؤلمة تؤثر على الأكیاس الصغیرة المملوءة بالسوائل والتي تسمى بالأجربة ووظیفتھا
حمایة العظام والأوتار والعضلات بالقرب من المفاصل" أو التھاب المفاصل (التھاب المفاصل
المصحوب عادة بألم، ورم و/أو تیبس)، ألم في الأطراف، ألم في الظھر، تشنجات عضلیة.
• وجود دم بالبول، تبول متكرر غیر طبیعي، ظھور نتائج غیر طبیعیة في فحوصات البول
(ارتفاع مستوى البروتینات في البول)، انخفاض كفاءة الكلى في القیام بوظائفھا بدقة.
• شعور بالتعب والإجھاد، ألم بالصدر، ضیق بالصدر.
• حصوات في المرارة أو في القنوات الصفراویة (تَحصّي صفراوي).
• زیادة مستوى الھرمون المحفز للغدة الدرقیة في الدم.
• تغیرات في كیمیاء الدم أو كمیة خلایا الدم أو الصفائح الدمویة (ظھورنتائج غیر طبیعیة في
فحوصات الدم).
• حصوات الكُلى.
• مشاكل في الانتصاب.
تأثیرات جانبیة نادرة (قد تُؤثر على ما یصل إلى مریض واحد من بین كل ۱٫۰۰۰ مریض):
• تلف العضلات، وقد تكون ھناك مضاعفات خطیرة لھذه الحالة. حیث قد تسبب ھذه الحالة
مشاكل في العضلات وعلى وجھ الخصوص إذا كنت تشعر في نفس الوقت بإعیاء أو ارتفاع في
درجة الحرارة فقد یكون ذلك بسبب شد العضلات بشكل غیر طبیعي. اتصل بالطبیب المعالج لك
فورًا إذا عانیت من آلام في العضلات، أو ألم أو ضعف.
• تورّم شدید في الطبقات الداخلیة للجلد، وعلى وجھ الخصوص حول الشفاه أو العینین أو
الأعضاء التناسلیة أو الیدین أو القدم أو اللسان، مع احتمال الإصابة بصعوبة مفاجئة في التنفس.
• حُمى شدیدة مصحوبة بطفح جلدي یشبھ الحصبة، تضخم العقد اللیمفاویة، تضخم الكبد، التھاب
الكبد (وصولًا إلى فشل الكبد)، ارتفاع عدد الخلایا البیضاء في الدم (كثرة كریات الدم البیضاء،
التي قد یصاحبھا فرط الكریات الحمضیة)
• احمرار الجلد (التھاب احمراري للجلد) طفح جلدي بأشكال مختلفة (مثل حكة مصحوبة ببقع
بیضاء، بثور، بثور تحتوي على صدید، احمرار الجلد، وطفح جلدي یشبھ الحصبة)، حمامي
منتشر بجمیع اجزاء الجسم، نَخَر (موت الأنسجة)، انحلال البشرة والأغشیة المخاطیة الفقاعیة،
ما قد یؤدي إلى الإصابة بتقشر وتقیح (متلازمة ستیفنز جونسون/ تَقَشُّرُ الأَنْسِجَةِ المُتَمَوِّتَةِ
البَشْرَوِیَّةِ التَّسَمُّمِيّ)
• عصبیة.
• شعور بالعطش.
• طنین في الأذنین.
• عدم وضوح الرؤیة، تغیرات في الرؤیة.
• تساقط الشعر.
• تقرح الفم.
• التھاب البنكریاس: الأعراض الشائعة ھي: ألم في البطن، غثیان، قيء.
• فرط التعرق.
• انخفاض الوزن، زیادة الشھیة، فقدان الشھیة اللاإرادي (فقدان الشھیة العصبي).
• تیبس العضلات و/أو المفاصل.
• انخفاض عدد خلایا الدم بصورة غیر طبیعیة (خلایا الدم الحمراء أو البیضاء أو الصفائح
الدمویة).
• شعور برغبة مُلحة في التبول.
• تغیرات أو انخفاض في كمیة البول نتیجة لالتھاب الكلى (التھاب الكلیة الخلالي).
• التھاب الكبد (التھاب الكبد الوبائي).
• اصفرار الجلد (یرقان).
• تلف الكبد.
• ارتفاع مستوى فسفوكیناز الكریاتین في الدم (وھو مؤشر على تلف العضلات).
الإبلاغ عن الآثار الجانبیة
إذا عانیت من أي من الآثار الجانبیة، تحدَّث إلى الطبیب المعالج لك أو الصیدلي الخاص بك.
یشمل ذلك أیّة آثار جانبیة مُحتمَلة غیر مُدرجة في ھذه النَّشرة. یمكنك المساعدة في توفیر
معلومات إضافیة حول أمان استخدام ھذا الدَّواء من خلال إبلاغك عن الآثار الجانبیة.
یحفظ بعیدًا عن متناول و مرأى الأطفال.
لا تستعمل ھذا الدَّواء بعد تاریخ انتھاء الصلاحیة المدون على العبوة.
یحفظ في درجة حرارة لا تزید عن ۳۰ درجة مئویة.
لا تتخلص من الأدویة عن طریق إلقائھا في میاه الصرف أو مع المخلفات المنزلیة.
استشر الصیدلي عن كیفیة التَّخلص من الأدویة التي لم تعد تستخدمھا. سوف تُساعد ھذه
الإجراءات في الحفاظ على البیئة.
محتویات عقار یوراجو
المادة الفعّالة ھي فیبوكسوستات
یحتوي كل قرص مغلف على ٤۰ ملجم أو ۸۰ ملجم من فیبوكسوستات.
المكونات الأخرى ھي:
لب القرص: مونوھیدرات اللاكتوز ، سلیلوز دقیق التَبلّور ، ھیدروكسي بروبیل السلیلوز ،
ثاني أكسید السیلیكون الغروي ، صودیوم كروسكارمیلوز وستیرات الماغنسیوم
غلاف القرص : كحول البولي فینیل ، میكروجیل / بي اي جي ، تلك ، كینولین أصفر ،
أزرق لامع ، أصفر غروب الشمس و ثاني أكسید التیتانیوم.
۰ ملجم أقراص لونھا أخضر ، مستدیرة الشكل محفور على أحد جانبیھا ™ یوراجو
“ وعلى الجانب الآخر ” 211 "JP"
"JP" ۸۰ ملجم أقراص لونھا أخضر ، مستطیلة الشكل محفور على أحد جانبیھا ™ یوراجو
“ وعلى الجانب الآخر ” 210
متوفرة في أقراص ٤۰ ملجم و ۸۰ ملجم في علب تحتوي كل علبة على ™ أقراص یوراجو
۳۰ قرص.
قد لا یتم تسویق جمیع العبوات.
شركة مصنع جمجوم للأدویة،
جدة، منطقة مكة، المملكة العربیة السعودیة.
+۹٦٦-۱۲- ھاتف: ٦۰۸۱۱۱۱
+۹٦٦-۱۲- فاكس: ٦۰۸۱۲۲۲
www.jamjoompharma.com : الموقع الإلكتروني
للإبلاغ عن أي أثار جانبیھ:
• المملكة العربیة السعودیة:
- المركز الوطني للتیقظ و السلامة الدوائیة
+۹٦٦-۱۱-۲۰٥- فاكس: ۷٦٦۲ o
للإتصال بالإدارة التنفیذیة للتیقظ وإدارة الأزمات. o
۲۳٤۰-۲۳٥٦- ۹٦٦ + , تحویلة: ۲۳۱۷ -۱۱- ھاتف: ۲۰۳۸۲۲۲ o
الخط الساخن للإبلاغ: ۱۹۹۹۹ o
npc.drug@sfda.gov.sa : برید إلكتروني o
www.sfda.gov.sa/npc : الموقع الالكتروني o
• دول الخلیج الأخرى:
- الرجاء الاتصال بالمؤسسات و الھیئات الوطنیة في كل دولة.
Treatment of chronic hyperuricaemia in conditions where urate deposition has already occurred (including a history, or presence of, tophus and/or gouty arthritis).
FEBUXOSTAT is indicated in adults.
The recommended oral dose of FEBUXOSTAT is 80 mg once daily without regard to food. If serum uric acid is > 6 mg/dL (357 μmol/L) after 2-4 weeks, FEBUXOSTAT 120 mg once daily may be considered.
FEBUXOSTAT works sufficiently quickly to allow retesting of the serum uric acid after 2 weeks. The therapeutic target is to decrease and maintain serum uric acid below 6 mg/dL (357 μmol/L).
Gout flare prophylaxis of at least 6 months is recommended (see section 4.4).
Elderly
No dose adjustment is required in the elderly (see section 5.2).
Renal impairment
The efficacy and safety have not been fully evaluated in patients with severe renal impairment (creatinine clearance <30 mL/min, see section 5.2).
No dose adjustment is necessary in patients with mild or moderate renal impairment.
Hepatic impairment
The efficacy and safety of febuxostat has not been studied in patients with severe hepatic impairment (Child Pugh Class C).
The recommended dose in patients with mild hepatic impairment is 80 mg. Limited information is available in patients with moderate hepatic impairment.
Paediatric population
The safety and the efficacy of FEBUXOSTAT in children aged below the age of 18 years have not been established. No data are available.
Method of administration
Oral use
FEBUXOSTAT should be taken by mouth and can be taken with or without food.
Cardio-vascular disorders
In patients with pre-existing major cardiovascular diseases (e.g. myocardial infarction, stroke or unstable angina), during the development of the product and in one post registrational study (CARES), a higher number of fatal cardiovascular events were observed with febuxostat when compared to allopurinol.
However, in a subsequent post registrational study (FAST), febuxostat was not inferior to allopurinol in the incidence of both fatal and non-fatal cardiovascular events.
Treatment of this patient group should be exercised cautiously and they should be monitored regularly.
For further details on cardiovascular safety of febuxostat refer to section 4.8 and section 5.1.
Medicinal product allergy / hypersensitivity
Rare reports of serious allergic/hypersensitivity reactions, including life-threatening Stevens-Johnson Syndrome, Toxic epidermal necrolysis and acute anaphylactic reaction/shock, have been collected in the post-marketing experience. In most cases, these reactions occurred during the first month of therapy with febuxostat. Some, but not all of these patients reported renal impairment and/or previous hypersensitivity to allopurinol. Severe hypersensitivity reactions, including Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) were associated with fever, haematological, renal or hepatic involvement in some cases.
Patients should be advised of the signs and symptoms and monitored closely for symptoms of allergic/hypersensitivity reactions (see section 4.8). Febuxostat treatment should be immediately stopped if serious allergic/hypersensitivity reactions, including Stevens-Johnson Syndrome, occur since early withdrawal is associated with a better prognosis. If patient has developed allergic/hypersensitivity reactions including Stevens-Johnson Syndrome and acute anaphylactic reaction/shock, febuxostat must not be re-started in this patient at any time.
Acute gouty attacks (gout flare)
Febuxostat treatment should not be started until an acute attack of gout has completely subsided. Gout flares may occur during initiation of treatment due to changing serum uric acid levels resulting in mobilization of urate from tissue deposits (see section 4.8 and 5.1). At treatment initiation with febuxostat flare prophylaxis for at least 6 months with an NSAID or colchicine is recommended (see section 4.2).
If a gout flare occurs during febuxostat treatment, it should not be discontinued. The gout flare should be managed concurrently as appropriate for the individual patient. Continuous treatment with febuxostat decreases frequency and intensity of gout flares.
Xanthine deposition
In patients in whom the rate of urate formation is greatly increased (e.g. malignant disease and its treatment, Lesch-Nyhan syndrome) the absolute concentration of xanthine in urine could, in rare cases, rise sufficiently to allow deposition in the urinary tract. As there has been no experience with febuxostat, its use in these populations is not recommended.
Mercaptopurine/azathioprine
Febuxostat use is not recommended in patients concomitantly treated with mercaptopurine/azathioprine as inhibition of xanthine oxidase by febuxostat may cause increased plasma concentrations of mercaptopurine/azathioprine that could result in severe toxicity.
Where the combination cannot be avoided, a reduction of the dose of mercaptopurine/azathioprine to the 20% or less of the previously prescribed dose is recommended in order to avoid possible haematological effects (see sections 4.5 and 5.3).
The patients should be closely monitored and the dose of mercaptopurine/azathioprine should be subsequently adjusted based on the evaluation of the therapeutic response and the onset of eventual toxic effects.
Organ transplant recipients
As there has been no experience in organ transplant recipients, the use of febuxostat in such patients is not recommended (see section 5.1).
Theophylline
Co-administration of febuxostat 80 mg and theophylline 400mg single dose in healthy subjects showed absence of any pharmacokinetic interaction (see section 4.5). Febuxostat 80 mg can be used in patients concomitantly treated with theophylline without risk of increasing theophylline plasma levels. No data is available for febuxostat 120 mg.
Liver disorders
During the combined phase 3 clinical studies, mild liver function test abnormalities were observed in patients treated with febuxostat (5.0%). Liver function test is recommended prior to the initiation of therapy with febuxostat and periodically thereafter based on clinical judgment (see section 5.1).
Thyroid disorders
Increased TSH values (>5.5 µIU/mL) were observed in patients on long-term treatment with febuxostat (5.5%) in the long term open label extension studies. Caution is required when febuxostat is used in patients with alteration of thyroid function (see section 5.1).
Lactose
Febuxostat tablets contain lactose. Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption should not take this medicine.
Sodium
This medicine contains less than 1 mmol sodium (23 mg) per tablet, that is to say essentially 'sodium-free'.
4.5 Interaction with other medicinal products and other forms of interaction
Mercaptopurine/azathioprine
On the basis of the mechanism of action of febuxostat on XO inhibition concomitant use is not recommended. Inhibition of XO by febuxostat may cause increased plasma concentrations of these drugs leading to toxicity. Drug interaction studies of febuxostat with drugs (except theophylline) that are metabolized by XO have not been performed in humans.
Modelling and simulation analysis of data from a pre-clinical study in rats indicates that, in case of concomitant administration with febuxostat, the dose of mercaptopurine/azathioprine should be reduced to 20% or less of the previously prescribed dose (see section 4.4 and 5.3).
Drug interaction studies of febuxostat with other cytotoxic chemotherapy have not been conducted. No data is available regarding the safety of febuxostat during other cytotoxic therapy.
Rosiglitazone/CYP2C8 substrates
Febuxostat was shown to be a weak inhibitor of CYP2C8 in vitro. In a study in healthy subjects, coadministration of 120 mg febuxostat QD with a single 4 mg oral dose of rosiglitazone had no effect on the pharmacokinetics of rosiglitazone and its metabolite N-desmethyl rosiglitazone, indicating that febuxostat is not a CYP2C8 enzyme inhibitor in vivo. Thus, co-administration of febuxostat with rosiglitazone or other CYP2C8 substrates is not expected to require any dose adjustment for those compounds.
Theophylline
An interaction study in healthy subjects has been performed with febuxostat to evaluate whether the inhibition of XO may cause an increase in the theophylline circulating levels as reported with other XO inhibitors. The results of the study showed that the co-administration of febuxostat 80 mg QD with theophylline 400 mg single dose has no effect on the pharmacokinetics or safety of theophylline. Therefore no special caution is advised when febuxostat 80 mg and theophylline are given concomitantly. No data is available for febuxostat 120 mg.
Naproxen and other inhibitors of glucuronidation
Febuxostat metabolism depends on Uridine Glucuronosyl Transferase (UGT) enzymes. Medicinal products that inhibit glucuronidation, such as NSAIDs and probenecid, could in theory affect the elimination of febuxostat. In healthy subjects concomitant use of febuxostat and naproxen 250 mg twice daily was associated with an increase in febuxostat exposure (Cmax 28%, AUC 41% and t1/2 26%). In clinical studies the use of naproxen or other NSAIDs/Cox-2 inhibitors was not related to any clinically significant increase in adverse events.
Febuxostat can be co-administered with naproxen with no dose adjustment of febuxostat or naproxen being necessary.
Inducers of glucuronidation
Potent inducers of UGT enzymes might possibly lead to increased metabolism and decreased efficacy of febuxostat. Monitoring of serum uric acid is therefore recommended 1-2 weeks after start of treatment with a potent inducer of glucuronidation. Conversely, cessation of treatment of an inducer might lead to increased plasma levels of febuxostat.
Colchicine/indometacin/hydrochlorothiazide/warfarin
Febuxostat can be co-administered with colchicine or indomethacin with no dose adjustment of febuxostat or the co-administered active substance being necessary.
No dose adjustment is necessary for febuxostat when administered with hydrochlorothiazide.
No dose adjustment is necessary for warfarin when administered with febuxostat. Administration of febuxostat (80 mg or 120 mg once daily) with warfarin had no effect on the pharmacokinetics of warfarin in healthy subjects. INR and Factor VII activity were also not affected by the co-administration of febuxostat.
Desipramine/CYP2D6 substrates
Febuxostat was shown to be a weak inhibitor of CYP2D6 in vitro. In a study in healthy subjects, 120 mg Febuxostat QD resulted in a mean 22% increase in AUC of desipramine, a CYP2D6 substrate indicating a potential weak inhibitory effect of febuxostat on the CYP2D6 enzyme in vivo. Thus, co-administration of febuxostat with other CYP2D6 substrates is not expected to require any dose adjustment for those compounds.
Antacids
Concomitant ingestion of an antacid containing magnesium hydroxide and aluminium hydroxide has been shown to delay absorption of febuxostat (approximately 1 hour) and to cause a 32% decrease in Cmax, but no significant change in AUC was observed. Therefore, febuxostat may be taken without regard to antacid use.
Pregnancy
Data on a very limited number of exposed pregnancies have not indicated any adverse effects of febuxostat on pregnancy or on the health of the foetus/new born child. Animal studies do not indicate direct or indirect harmful effects with respect to pregnancy, embryonal/foetal development or parturition (see section 5.3). The potential risk for human is unknown. Febuxostat should not be used during pregnancy.
Breastfeeding
It is unknown whether febuxostat is excreted in human breast milk. Animal studies have shown excretion of this active substance in breast milk and an impaired development of suckling pups. A risk to a suckling infant cannot be excluded. Febuxostat should not be used while breastfeeding.
Fertility
In animals, reproduction studies up to 48 mg/kg/day showed no dose-dependent adverse effects on fertility (see section 5.3). The effect of Febuxostat on human fertility is unknown.
Somnolence, dizziness, paraesthesia and blurred vision have been reported with the use of Febuxostat. Patients should exercise caution before driving, using machinery or participating in dangerous activities until they are reasonably certain that FEBUXOSTAT does not adversely affect performance.
Summary of the safety profile
The most commonly reported adverse reactions in clinical trials (4,072 subjects treated at least with a dose from 10 mg to 300 mg), post-authorisation safety studies (FAST study: 3001 subjects treated at least with a dose from 80 mg to 120 mg) and post-marketing experience are gout flares, liver function abnormalities, diarrhoea, nausea, headache, dizziness, dyspnoea, rash, pruritus,arthralgia, myalgia, pain in extremity, oedema and fatigue. These adverse reactions were mostly mild or moderate in severity. Rare serious hypersensitivity reactions to febuxostat, some of which were associated to systemic symptoms, and rare events of sudden cardiac death, have occurred in the post-marketing experience.
Tabulated list of adverse reactions
Common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100) and rare (≥1/10,000 to <1/1,000) adverse reactions occurring in patients treated with febuxostat are listed below.
Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness.
Table 1: Adverse reactions in combined phase 3, long-term extension studies, post-authorisation safety studies and post-marketing experience
Blood and lymphatic system disorders | Rare Pancytopenia, thrombocytopenia, agranulocytosis*, anaemia# |
Immune system disorders | Rare Anaphylactic reaction*, drug hypersensitivity* |
Endocrine disorders | Uncommon Blood thyroid stimulating hormone increased, hypothyroidism# |
Eye disorders | Uncommon Blurred vision Rare Retinal artery occlusion# |
Metabolism and nutrition disorders | Common*** Gout flares Uncommon Diabetes mellitus, hyperlipidemia, decrease appetite, weight increase Rare Weight decrease, increase appetite, anorexia |
Psychiatric disorders | Uncommon Libido decreased, insomnia Rare Nervousness, depressed mood#, sleep disorder# |
Nervous system disorders | Common Headache, dizziness Uncommon Paraesthesia, hemiparesis, somnolence, lethargy# altered taste, hypoaesthesia, hyposmia Rare Ageusia#, burning sensation# |
Ear and labyrinth disorders | Uncommon Tinnitus Rare Vertigo# |
Cardiac disorders | Uncommon Atrial fibrillation, palpitations, ECG abnormal, arrhythmia# Rare Sudden cardiac death* |
Vascular disorders | Uncommon Hypertension, flushing, hot flush Rare Circulatory collapse# |
Respiratory system disorders | Common Dyspnoea Uncommon Bronchitis, upper respiratory tract infection, lower respiratory tract infection#, cough, rhinorrhoea# Rare Pneumonia# |
Gastrointestinal disorders | Common Diarrhoea**, nausea Uncommon: Abdominal pain, abdominal pain upper #, abdominal distension, gastro-oesophageal reflux disease, vomiting, dry mouth, dyspepsia, constipation, frequent stools, flatulence, gastrointestinal discomfort, mouth ulceration, lip swelling #, pancreatitis Rare Gastrointestinal perforation #, stomatitis# |
Hepato-biliary disorders | Common Liver function abnormalities** Uncommon Cholelithiasis Rare Hepatitis, jaundice*, liver injury*, cholecystitis# |
Skin and subcutaneous tissue disorders | Common Rash (including various types of rash reported with lower frequencies, see below), pruritus Uncommon Dermatitis, urticaria, skin discolouration, skin lesion, petechiae, rash macular, rash maculopapular, rash papular, hyperhidrosis, alopecia, eczema #, erythema, night sweats #, psoriasis#, rash pruritic# Rare Toxic epidermal necrolysis*, Stevens-Johnson Syndrome*, angioedema*, drug reaction with eosinophilia and systemic symptoms*, generalized rash (serious)*, exfoliative rash, rash follicular, rash vesicular, rash pustular, rash erythematous, rash morbillifom |
Musculoskeletal and connective tissue disorders | Common Arthralgia, myalgia, pain in extremity# Uncommon Arthritis, musculoskeletal pain, muscle weakness, muscle spasm, muscle tightness, bursitis, joint swelling #, back pain #, musculoskeletal stiffness#, joint stiffness Rare Rhabdomyolysis*, rotator cuff syndrome #, polymyalgia rheumatica# |
Renal and urinary disorders | Uncommon Renal failure, nephrolithiasis, haematuria, pollakiuria, proteinuria, micturition urgency, urinary tract infection# Rare Tubulointerstitial nephritis* |
Reproductive system and breast disorder | Uncommon Erectile dysfunction |
General disorders and administration site conditions | Common Oedema, Fatigue Uncommon Chest pain, chest discomfort, pain #, malaise# Rare Thirst, feeling hot# |
Investigations | Uncommon Blood amylase increase, platelet count decrease, WBC decrease, lymphocyte count decrease, blood creatine increase, blood creatinine increase, haemoglobin decrease, blood urea increase, blood triglycerides increase, blood cholesterol increase, haematocritic decrease, blood lactate dehydrogenase increased, blood potassium increase, INR increased# Rare Blood glucose increase, activated partial thromboplastin time prolonged, red blood cell count decrease, blood alkaline phosphatase increase, blood creatine phosphokinase increase* |
Injury, poisoning and procedural complications | Uncommon Contusion# |
* Adverse reactions coming from post-marketing experience
** Treatment-emergent non-infective diarrhoea and abnormal liver function tests in the combined Phase 3 studies are more frequent in patients concomitantly treated with colchicine.
*** See section 5.1 for incidences of gout flares in the individual Phase 3 randomized controlled studies.
# Adverse reactions coming from post-authorisation safety studies
Description of selected adverse reactions
Rare serious hypersensitivity reactions to febuxostat, including Stevens-Johnson Syndrome, Toxic epidermal necrolysis and anaphylactic reaction/shock, have occurred in the post-marketing experience. Stevens-Johnson Syndrome and Toxic epidermal necrolysis are characterised by progressive skin rashes associated with blisters or mucosal lesions and eye irritation. Hypersensitivity reactions to febuxostat can be associated to the following symptoms: skin reactions characterised by infiltrated maculopapular eruption, generalised or exfoliative rashes, but also skin lesions, facial oedema, fever, haematologic abnormalities such as thrombocytopenia and eosinophilia, and single or multiple organ involvement (liver and kidney including tubulointerstitial nephritis) (see section 4.4).
Gout flares were commonly observed soon after the start of treatment and during the first months. Thereafter, the frequency of gout flare decreases in a time-dependent manner. Gout flare prophylaxis is recommended (see section 4.2 and 4.4).
Patients with an overdose should be managed by symptomatic and supportive care
Pharmacotherapeutic group: Antigout preparation, preparations inhibiting uric acid production, ATC code: M04AA03
Mechanism of action
Uric acid is the end product of purine metabolism in humans and is generated in the cascade of hypoxanthine → xanthine → uric acid. Both steps in the above transformations are catalyzed by xanthine oxidase (XO). Febuxostat is a 2-arylthiazole derivative that achieves its therapeutic effect of decreasing serum uric acid by selectively inhibiting XO. Febuxostat is a potent, non-purine selective inhibitor of XO (NP-SIXO) with an in vitro inhibition Ki value less than one nanomolar. Febuxostat has been shown to potently inhibit both the oxidized and reduced forms of XO. At therapeutic concentrations febuxostat does not inhibit other enzymes involved in purine or pyrimidine metabolism, namely, guanine deaminase, hypoxanthine guanine phosphoribosyltransferase, orotate phosphoribosyltransferase, orotidine monophosphate decarboxylase or purine nucleoside phosphorylase.
Clinical efficacy and safety
The efficacy of Febuxostat was demonstrated in three Phase 3 pivotal studies (the two pivotal APEX and FACT studies, and the additional CONFIRMS study described below) that were conducted in 4101 patients with hyperuricaemia and gout. In each phase 3 pivotal study, Febuxostat demonstrated superior ability to lower and maintain serum uric acid levels compared to allopurinol. The primary efficacy endpoint in the APEX and FACT studies was the proportion of patients whose last 3 monthly serum uric acid levels were < 6.0 mg/dL (357 µmol/L). In the additional phase 3 CONFIRMS study, for which results became available after the marketing authorisation for Febuxostat was first issued, the primary efficacy endpoint was the proportion of patients whose serum urate level was < 6.0 mg/dL at the final visit. No patients with organ transplant have been included in these studies (see section 4.2).
APEX Study: The Allopurinol and Placebo-Controlled Efficacy Study of Febuxostat (APEX) was a Phase 3, randomized, double-blind, multicenter, 28-week study. One thousand and seventy-two (1072) patients were randomized: placebo (n=134), Febuxostat 80 mg QD (n=267), Febuxostat 120 mg QD (n=269), Febuxostat 240 mg QD (n=134) or allopurinol (300 mg QD [n=258] for patients with a baseline serum creatinine ≤1.5 mg/dL or 100 mg QD [n=10] for patients with a baseline serum creatinine >1.5 mg/dL and ≤2.0 mg/dL). Two hundred and forty mg febuxostat (2 times the recommended highest dose) was used as a safety evaluation dose.
The APEX study showed statistically significant superiority of both the Febuxostat 80 mg QD and the Febuxostat 120 mg QD treatment arms versus the conventionally used doses of allopurinol 300 mg (n = 258) /100 mg (n = 10) treatment arm in reducing the sUA below 6 mg/dL (357 µmol/L) (see Table 2 and Figure 1).
FACT Study: The Febuxostat Allopurinol Controlled Trial (FACT) Study was a Phase 3, randomized, double-blind, multicenter, 52-week study. Seven hundred sixty (760) patients were randomized: Febuxostat 80 mg QD (n=256), Febuxostat 120 mg QD (n=251), or allopurinol 300 mg QD (n=253).
The FACT study showed the statistically significant superiority of both Febuxostat 80 mg and Febuxostat 120 mg QD treatment arms versus the conventionally used dose of allopurinol 300 mg treatment arm in reducing and maintaining sUA below 6 mg/dL (357 µmol/L).
Table 2 summarises the primary efficacy endpoint results:
Table 2
Proportion of Patients with Serum Uric Acid Levels <6.0 mg/dL (357 µmol/L)
Last Three Monthly Visits
Study | Febuxostat 80 mg QD | Febuxostat 120 mg QD | Allopurinol 300 / 100 mg QD1 |
APEX (28 weeks) | 48% * (n=262) | 65% *, # (n=269) | 22% (n=268) |
FACT (52 weeks) | 53%* (n=255) | 62%* (n=250) | 21% (n=251) |
Combined Results | 51%* (n=517) | 63%*, # (n=519) | 22% (n=519) |
1 results from subjects receiving either 100 mg QD (n=10: patients with serum creatinine >1.5 and ≤2.0 mg/dL) or 300 mg QD (n=509) were pooled for analyses. * p < 0.001 vs allopurinol, # p < 0.001 vs 80 mg | |||
The ability of Febuxostat to lower serum uric acid levels was prompt and persistent. Reduction in serum uric acid level to <6.0 mg/dL (357 µmol/L) was noted by the Week 2 visit and was maintained throughout treatment. The mean serum uric acid levels over time for each treatment group from the two pivotal Phase 3 studies are shown in Figure 1.
Figure 1 Mean Serum Uric Acid Levels in Combined Pivotal Phase 3 Studies
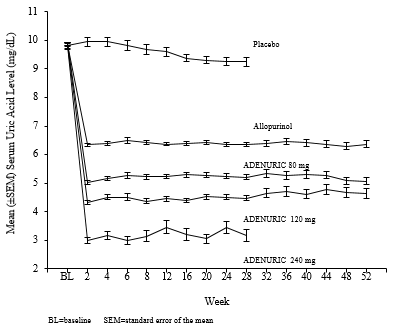
Note: 509 patients received allopurinol 300 mg QD; 10 patients with serum creatinine >1.5 and ≤2.0 mg/dL were dosed with 100 mg QD. (10 patients out of 268 in APEX study).
240 mg febuxostat was used to evaluate the safety of febuxostat at twice the recommended highest dose.
CONFIRMS Study: The CONFIRMS study was a Phase 3, randomized, controlled, 26-week study to evaluate the safety and efficacy of febuxostat 40 mg and 80 mg, in comparison with allopurinol 300 mg or 200 mg, in patients with gout and hyperuricaemia. Two thousand and two hundred-sixty nine (2269) patients were randomized: Febuxostat 40 mg QD (n=757), Febuxostat 80 mg QD (n=756), or allopurinol 300/200 mg QD (n=756). At least 65% of the patients had mild-moderate renal impairment (with creatinine clearance of 30-89 mL/min). Prophylaxis against gout flares was obligatory over the 26-week period.
The proportion of patients with serum urate levels of < 6.0 mg/dL (357 µmol/L) at the final visit, was 45% for 40 mg febuxostat, 67% for febuxostat 80 mg and 42% for allopurinol 300/200 mg, respectively.
Primary endpoint in the sub-group of patients with renal impairment
The APEX Study evaluated efficacy in 40 patients with renal impairment (i.e., baseline serum creatinine > 1.5 mg/dL and ≤2.0 mg/dL). For renally impaired subjects who were randomized to allopurinol, the dose was capped at 100 mg QD. Febuxostat achieved the primary efficacy endpoint in 44% (80 mg QD), 45% (120 mg QD), and 60% (240 mg QD) of patients compared to 0% in the allopurinol 100 mg QD and placebo groups.
There were no clinically significant differences in the percent decrease in serum uric acid concentration in healthy subjects irrespective of their renal function (58% in the normal renal function group and 55% in the severe renal dysfunction group).
An analysis in patients with gout and renal impairment was prospectively defined in the CONFIRMS study, and showed that febuxostat was significantly more efficacious in lowering serum urate levels to < 6 mg/dL compared to allopurinol 300 mg/200 mg in patients who had gout with mild to moderate renal impairment (65% of patients studied).
Primary endpoint in the sub group of patients with sUA ≥ 10 mg/dL
Approximately 40% of patients (combined APEX and FACT) had a baseline sUA of ≥ 10 mg/dL. In this subgroup Febuxostat achieved the primary efficacy endpoint (sUA < 6.0 mg/dL at the last 3 visits) in 41% (80 mg QD), 48% (120 mg QD), and 66% (240 mg QD) of patients compared to 9% in the allopurinol 300 mg/100 mg QD and 0 % in the placebo groups.
In the CONFIRMS study, the proportion of patients achieving the primary efficacy endpoint (sUA < 6.0 mg/dL at the final visit) for patients with a baseline serum urate level of ≥ 10 mg/dL treated with febuxostat 40 mg QD was 27% (66/249), with febuxostat 80 mg QD 49% (125/254) and with allopurinol 300 mg/200 mg QD 31% (72/230), respectively.
Clinical Outcomes: proportion of patients requiring treatment for a gout flare
APEX study: During the 8-week prophylaxis period, a greater proportion of subjects in the febuxostat 120 mg (36%) treatment group required treatment for gout flare compared to febuxostat 80 mg (28%), allopurinol 300 mg (23%) and placebo (20%). Flares increased following the prophylaxis period and gradually decreased over time. Between 46% and 55% of subjects received treatment for gout flares from Week 8 and Week 28. Gout flares during the last 4 weeks of the study (Weeks 24-28) were observed in 15% (febuxostat 80, 120 mg), 14% (allopurinol 300 mg) and 20% (placebo) of subjects.
FACT study: During the 8-week prophylaxis period, a greater proportion of subjects in the febuxostat 120 mg (36%) treatment group required treatment for a gout flare compared to both the febuxostat 80 mg (22%) and allopurinol 300 mg (21%) treatment groups. After the 8-week prophylaxis period, the incidences of flares increased and gradually decreased over time (64% and 70% of subjects received treatment for gout flares from Week 8-52). Gout flares during the last 4 weeks of the study (Weeks 49-52) were observed in 6-8% (febuxostat 80 mg, 120 mg) and 11% (allopurinol 300 mg) of subjects.
The proportion of subjects requiring treatment for a gout flare (APEX and FACT Study) was numerically lower in the groups that achieved an average post-baseline serum urate level <6.0 mg/dL, <5.0 mg/dL, or <4.0 mg/dL compared to the group that achieved an average post-baseline serum urate level ≥6.0 mg/dL during the last 32 weeks of the treatment period (Week 20-Week 24 to Week 49 - 52 intervals).
During the CONFIRMS study, the percentages of patients who required treatment for gout flares (Day 1 through Month 6) were 31% and 25% for the febuxostat 80 mg and allopurinol groups, respectively. No difference in the proportion of patients requiring treatment for gout flares was observed between the febuxostat 80 mg and 40 mg groups.
Long-term, open label extension Studies
EXCEL Study (C02-021): The Excel study was a three years Phase 3, open label, multicenter, randomised, allopurinol-controlled, safety extension study for patients who had completed the pivotal Phase 3 studies (APEX or FACT). A total of 1,086 patients were enrolled: Febuxostat 80 mg QD (n=649), Febuxostat 120 mg QD (n=292) and allopurinol 300/100 mg QD (n=145). About 69 % of patients required no treatment change to achieve a final stable treatment. Patients who had 3 consecutive sUA levels >6.0 mg/dL were withdrawn.
Serum urate levels were maintained over time (i.e. 91% and 93% of patients on initial treatment with febuxostat 80 mg and 120 mg, respectively, had sUA <6 mg/dL at Month 36).
Three years data showed a decrease in the incidence of gout flares with less than 4% of patients requiring treatment for a flare (i.e. more than 96% of patients did not require treatment for a flare) at Month 16-24 and at Month 30-36.
46% and 38%, of patients on final stable treatment of febuxostat 80 or 120 mg QD, respectively, had complete resolution of the primary palpable tophus from baseline to the Final Visit.
FOCUS Study (TMX-01-005) was a 5 years Phase 2, open-label, multicenter, safety extension study for patients who had completed the febuxostat 4 weeks of double blind dosing in study TMX-00-004. 116 patients were enrolled and received initially febuxostat 80 mg QD. 62% of patients required no dose adjustment to maintain sUA <6 mg/dL and 38% of patients required a dose adjustment to achieve a final stable dose.
The proportion of patients with serum urate levels of <6.0 mg/dL (357 µmol/L) at the final visit was greater than 80% (81-100%) at each febuxostat dose.
During the phase 3 clinical studies, mild liver function test abnormalities were observed in patients treated with febuxostat (5.0%). These rates were similar to the rates reported on allopurinol (4.2%) (see section 4.4). Increased TSH values (>5.5 µIU/mL) were observed in patients on long-term treatment with febuxostat (5.5%) and patients with allopurinol (5.8%) in the long term open label extension studies (see section 4.4).
Post Marketing long term studies
CARES Study was a multicenter, randomized, double-blind, non inferiority trial comparing CV outcomes with febuxostat versus allopurinol in patients with gout and a history of major CV disease including MI, hospitalization for unstable angina, coronary or cerebral revascularization procedure, stroke, hospitalized transient ischemic attack, peripheral vascular disease, or diabetes mellitus with evidence of microvascular or macrovascular disease. To achieve sUA less than 6 mg/dL, the dose of febuxostat was titrated from 40 mg up to 80 mg (regardless of renal function) and the dose of allopurinol was titrated in 100 mg increments from 300 to 600 mg in patients with normal renal function and mild renal impairment and from 200 to 400 mg in patients with moderate renal impairment.
The primary endpoint in CARES was the time to first occurrence of MACE, a composite of non-fatal MI, non-fatal stroke, CV death and unstable angina with urgent coronary revascularization.
The endpoints (primary and secondary) were analysed according to the intention-to-treat (ITT) analysis including all subjects who were randomized and received at least one dose of double-blind study medication.
Overall 56.6% of patients discontinued trial treatment prematurely and 45% of patients did not complete all trial visits.
In total, 6,190 patients were followed for a median of 32 months and the median duration of exposure was 728 days for patients in febuxostat group (n 3098) and 719 days in allopurinol group (n 3092).
The primary MACE endpoint occurred at similar rates in the febuxostat and allopurinol treatment groups (10.8% vs. 10.4% of patients, respectively; hazard ratio [HR] 1.03; two-sided repeated 95% confidence interval [CI] 0.89-1.21).
In the analysis of the individual components of MACE, the rate of CV deaths was higher with febuxostat than allopurinol (4.3% vs. 3.2% of patients; HR 1.34; 95% CI 1.03-1.73). The rates of the other MACE events were similar in the febuxostat and allopurinol groups, i.e. non-fatal MI (3.6% vs. 3.8% of patients; HR 0.93; 95% CI 0.72-1.21), non-fatal stroke (2.3% vs. 2.3% of patients; HR 1.01; 95% CI 0.73-1.41) and urgent revascularization due to unstable angina (1.6% vs. 1.8% of patients; HR 0.86; 95% CI 0.59-1.26). The rate of all-cause mortality was also higher with febuxostat than allopurinol (7.8% vs. 6.4% of patients; HR 1.22; 95% CI 1.01-1.47), which was mainly driven by the higher rate of CV deaths in that group (see section 4.4).
Rates of adjudicated hospitalization for heart failure, hospital admissions for arrhythmias not associated with ischemia, venous thromboembolic events and hospitalization for transient ischemic attacks were comparable for febuxostat and allopurinol.
FAST study was a prospective, randomised, open-label, blinded-endpoint study comparing the CV safety profile of febuxostat versus allopurinol in patients with chronic hyperuricaemia (in conditions where urate deposition had already occurred) and CV risk factors (i.e. patients 60 years or older and with at least one other CV risk factor). Eligible patients received allopurinol treatment prior to randomization, and dose adjustments were required when needed, according to clinical judgement, EULAR recommendations and the approved posology. At the end of the allopurinol lead-in phase, patients with a sUA level of <0.36 mmol/L (<6 mg/dL) or receiving the maximum tolerated dose or the maximum licensed dose of allopurinol were randomised in a 1:1 ratio to receive either febuxostat or allopurinol treatment. The primary endpoint of the study FAST was the time to the first occurrence of any event included in the Antiplatelet Trialists' Collaborative (APTC) composite endpoint, which included: i) hospitalisation for non-fatal MI/biomarker positive acute coronary syndrome (ACS); ii) non-fatal stroke; iii) death due to a CV event. The primary analysis was based on the on-treatment (OT) approach.
Overall, 6,128 patients were randomized, 3063 to febuxostat and 3065 to allopurinol.
In the primary OT analysis, febuxostat was non-inferior to allopurinol in the incidence of the primary endpoint, which occurred in 172 patients (1.72/100 patient years) on febuxostat compared to 241 patients (2.05/100 patient years) on allopurinol, with an adjusted HR 0.85 (95% CI: 0.70, 1.03), p<0.001. The OT analysis for the primary endpoint in the subgroup of patients with a history of MI, stroke or ACS showed no significant difference between treatment groups: there were 65 (9.5%) patients with events in the febuxostat group and 83 (11.8%) patients with events in the allopurinol group; adjusted HR 1.02 (95% CI: 0.74-1.42); p=0.202.
Treatment with febuxostat was not associated with an increase in CV death or all-cause death, overall or in the subgroup of patients with a baseline history of MI, stroke or ACS. Overall, there were fewer deaths in the febuxostat group (62 CV deaths and 108 all-cause deaths), than in the allopurinol group (82 CV deaths and 174 all-cause deaths).
There was a greater reduction in uric acid levels on febuxostat treatment compared to allopurinol treatment.
In healthy subjects, maximum plasma concentrations (Cmax) and area under the plasma concentration time curve (AUC) of febuxostat increased in a dose proportional manner following single and multiple doses of 10 mg to 120 mg. For doses between 120 mg and 300 mg, a greater than dose proportional increase in AUC is observed for febuxostat. There is no appreciable accumulation when doses of 10 mg to 240 mg are administered every 24 hours. Febuxostat has an apparent mean terminal elimination half-life (t1/2) of approximately 5 to 8 hours.
Population pharmacokinetic/pharmacodynamic analyses were conducted in 211 patients with hyperuricaemia and gout, treated with FEBUXOSTAT 40-240 mg QD. In general, febuxostat pharmacokinetic parameters estimated by these analyses are consistent with those obtained from healthy subjects, indicating that healthy subjects are representative for pharmacokinetic/pharmacodynamic assessment in the patient population with gout.
Absorption
Febuxostat is rapidly (tmax of 1.0-1.5 h) and well absorbed (at least 84%). After single or multiple oral 80 and 120 mg once daily doses, Cmax is approximately 2.8-3.2 μg/mL, and 5.0-5.3 μg/mL, respectively. Absolute bioavailability of the febuxostat tablet formulation has not been studied.
Following multiple oral 80 mg once daily doses or a single 120 mg dose with a high fat meal, there was a 49% and 38% decrease in Cmax and a 18% and 16% decrease in AUC, respectively. However, no clinically significant change in the percent decrease in serum uric acid concentration was observed where tested (80 mg multiple dose). Thus, FEBUXOSTAT may be taken without regard to food.
Distribution
The apparent steady state volume of distribution (Vss/F) of febuxostat ranges from 29 to 75 L after oral doses of 10-300 mg. The plasma protein binding of febuxostat is approximately 99.2%, (primarily to albumin), and is constant over the concentration range achieved with 80 and 120 mg doses. Plasma protein binding of the active metabolites ranges from about 82% to 91%.
Biotransformation
Febuxostat is extensively metabolized by conjugation via uridine diphosphate glucuronosyltransferase (UDPGT) enzyme system and oxidation via the cytochrome P450 (CYP) system. Four pharmacologically active hydroxyl metabolites have been identified, of which three occur in plasma of humans. In vitro studies with human liver microsomes showed that those oxidative metabolites were formed primarily by CYP1A1, CYP1A2, CYP2C8 or CYP2C9 and febuxostat glucuronide was formed mainly by UGT 1A1, 1A8, and 1A9.
Elimination
Febuxostat is eliminated by both hepatic and renal pathways. Following an 80 mg oral dose of 14C-labeled febuxostat, approximately 49% of the dose was recovered in the urine as unchanged febuxostat (3%), the acyl glucuronide of the active substance (30%), its known oxidative metabolites and their conjugates (13%), and other unknown metabolites (3%). In addition to the urinary excretion, approximately 45% of the dose was recovered in the faeces as the unchanged febuxostat (12%), the acyl glucuronide of the active substance (1%), its known oxidative metabolites and their conjugates (25%), and other unknown metabolites (7%).
Renal impairment
Following multiple doses of 80 mg of FEBUXOSTAT in patients with mild, moderate or severe renal impairment, the Cmax of febuxostat did not change, relative to subjects with normal renal function. The mean total AUC of febuxostat increased by approximately 1.8-fold from 7.5 μg·h/mL in the normal renal function group to 13.2 μg.h/mL in the severe renal dysfunction group. The Cmax and AUC of active metabolites increased up to 2- and 4-fold, respectively. However, no dose adjustment is necessary in patients with mild or moderate renal impairment.
Hepatic impairment
Following multiple doses of 80 mg of FEBUXOSTAT in patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment, the Cmax and AUC of febuxostat and its metabolites did not change significantly compared to subjects with normal hepatic function. No studies have been conducted in patients with severe hepatic impairment (Child-Pugh Class C).
Effects in non-clinical studies were generally observed at exposures in excess of the maximum human exposure.
Pharmacokinetic modelling and simulation of rat data suggests that, when co-administered with febuxostat, the clinical dose of mercaptopurine/azathioprine should be reduced to 20% or less of the previously prescribed dose in order to avoid possible haematological effects (see section 4.4 and 4.5).
Carcinogenesis, mutagenesis, impairment of fertility
In male rats, a statistically significant increase in urinary bladder tumours (transitional cell papilloma and carcinoma) was found only in association with xanthine calculi in the high dose group, at approximately 11 times human exposure. There was no significant increase in any other tumour type in either male or female mice or rats. These findings are considered a consequence of species specific purine metabolism and urine composition and of no relevance to clinical use.
A standard battery of test for genotoxicity did not reveal any biologically relevant genotoxic effects for febuxostat.
Febuxostat at oral doses up to 48 mg/kg/day was found to have no effect on fertility and reproductive performance of male and female rats.
There was no evidence of impaired fertility, teratogenic effects, or harm to the foetus due to febuxostat. There was high dose maternal toxicity accompanied by a reduction in weaning index and reduced development of offspring in rats at approximately 4.3 times human exposure. Teratology studies, performed in pregnant rats at approximately 4.3 times and pregnant rabbits at approximately 13 times human exposure did not reveal any teratogenic effects.
Microcrystalline Cellulose PH 101
Lactose Monohydrate
Hydroxypropyl Cellulose LF
Croscarmellose Sodium
Colloidal Silicon Dioxide-Aerosil 200
Magnesium Stearate
Opadry II Green 85F210131
Purified Water
Not applicable.
Do not store above 30 °C
3X10’s blister pack
Any unused product should be disposed of in accordance with local requirements.
